Methods
Our lab has developed two carefully-designed and validated machine learning and statistical methods for cell type-specific marker gene selection and cell type matching of single cell transcriptomic profiling data. NS-Forest is a random forest machine learning-based algorithm that selects the minimum set of necessary and sufficient (NS) marker genes to characterize the defining distinctions of different cell types. FR-Match is a graph theoretical approach to identify common and novel cell types using the multivariate nonparametric statistical test, Friedman-Rafsky (FR) test. These methods form the backbone of the Celligrate workflow.
NS-Forest
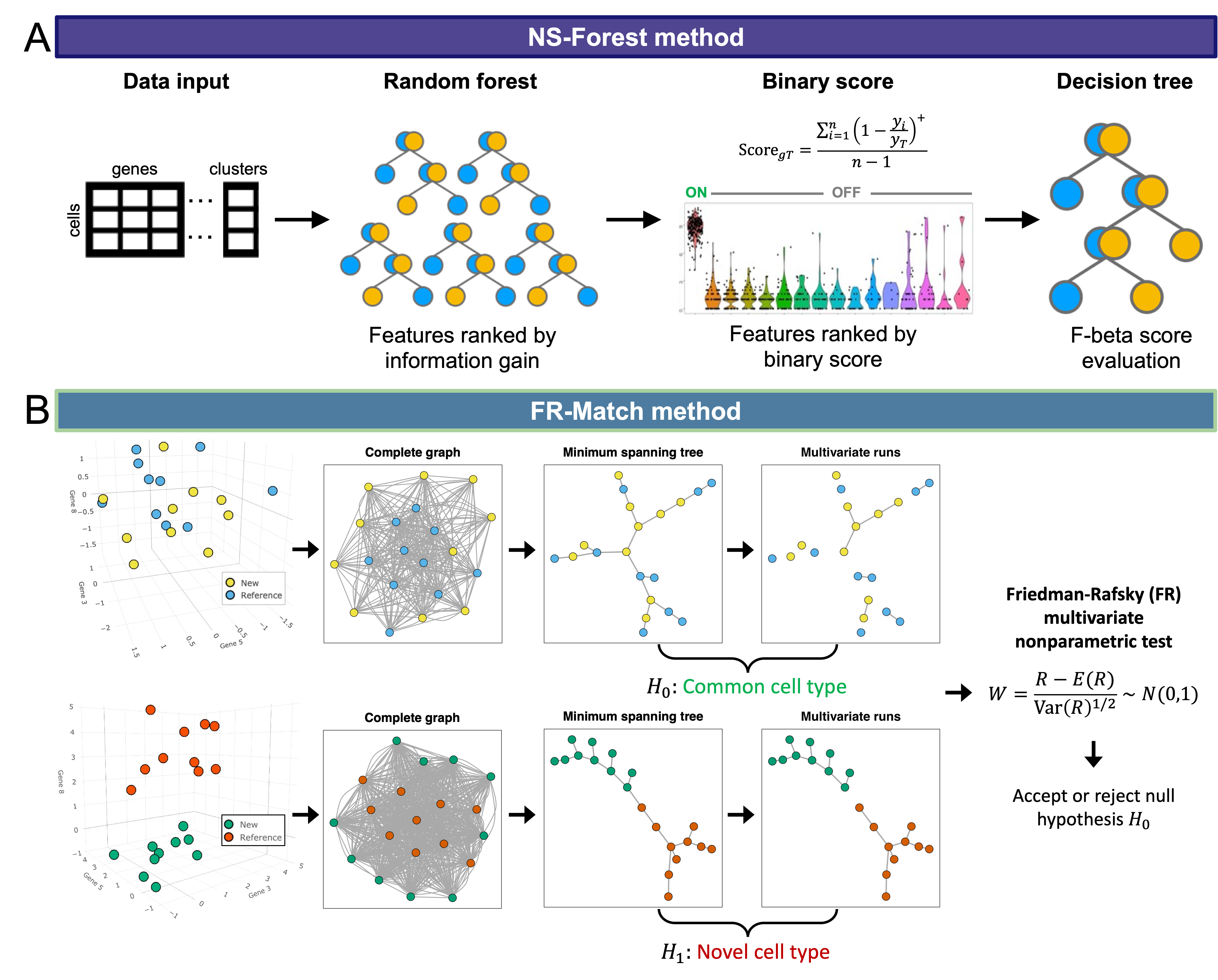
The NS-Forest algorithm (Figure A) takes the clustered cell-by-gene expression matrix and ranks the feature importance by the Gini index values calculated in the random forests classifiers. The Gini index-sorted features are great for machines to build quantitative classifiers, but they may not be the best candidates for certain biological and experimental use cases that require on-off binary signals in downstream applications, e.g., designing probes for PCR or multiplex FISH assays. We constructed a novel binary score based on the median expression levels within and across clusters to preferentially rank the positively-expressed binary gene features higher. The double-ranked feature list is used to determine the minimum set of features that gives the best classification accuracy in decision tree classifiers measured by the F-beta measure.
FR-Match
FR-Match is a novel application of the Friedman-Rafsky (FR) nonparametric statistical test for multivariate data comparison using minimum spanning tree graph analysis applied to single cell clustering results. FR-Match takes clustered gene expression matrices from query and reference experiments and returns the FR statistic with p-value as evidence that two cell clusters are matched or not, i.e., sharing a common gene expression profile pattern. In brief, the algorithm automatically detects whether cell clusters are intermixed with each other or segregated from each other in the selected feature space based on their joint minimum structure tree topology (Figure B).
Normalization
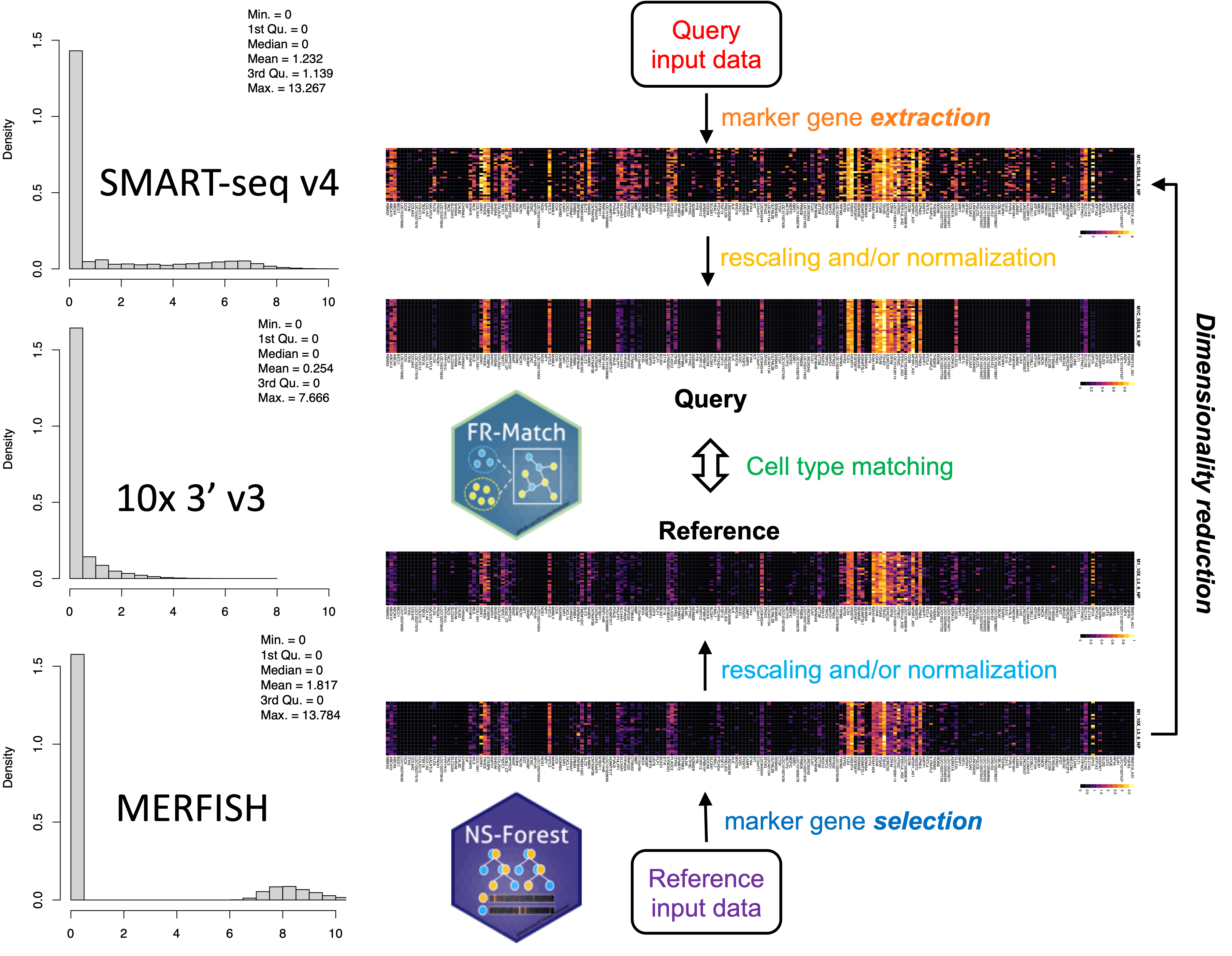
Single cell sequencing experiments differ in many aspects, including the different single cell technology platforms used prior to sequencing. These platforms, e.g., SMART-seq v4, 10x Chromium 3’ v3, MERFISH spatial transcriptomics, etc., vary largely by sensitivity and detection rate. We have recently developed an approach for expression data rescaling and normalization that denoises the expression data and allows for the use of FR-Match to match cell type clusters across different platforms.